Torres, T.; Gouloumis, A.; Sanchez-Garcia, D.; Jayawickramarajah, J.; Seitz, W.; Guldi, D. M.; Sessler, J. L. Photophysical characterization of a cytidine–guanosine tethered phthalocyanine–fullerene dyad. Chemical Communications 2007, 292-294.
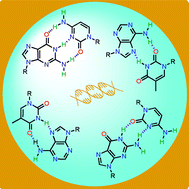
A new non-covalent electron transfer model system, based on the use of cytidine–guanosine hydrogen bonding interactions, is described that incorporates a phthalocyanine photodonor and a C60 fullerene acceptor.